Arch. Endocrinol. Metab. 2026;70(special issue 1): e250428
Clinical approach to the male with delayed puberty
DOI: 10.20945/2359-4292-2025-0428
ABSTRACT
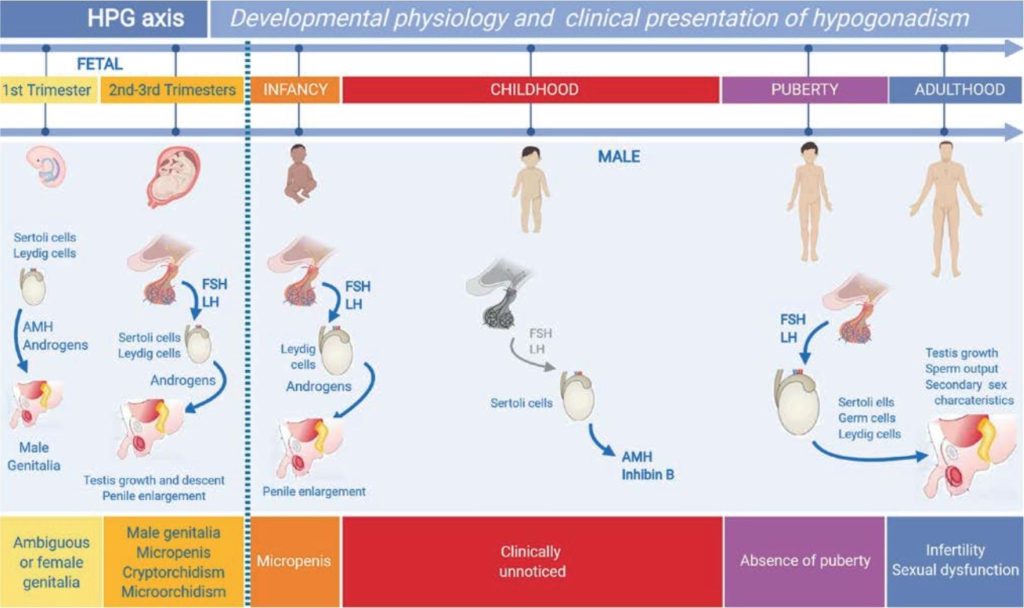
Disorders of pubertal onset and progression are a common cause for referral to paediatric endocrinologists, with delayed puberty in males being particularly frequent. Pubertal development depends on the hypothalamic-pituitary-testicular (HPT) axis, which is established during fetal life and undergoes distinct phases: fetal androgen production, postnatal “minipuberty”, and reactivation during adolescence. Key regulators include GnRH neurons, Sertoli and Leydig cells, and biomarkers such as AMH, inhibin B, testosterone and INSL3. Puberty is marked clinically by testicular enlargement beyond 4 mL, usually at a median age of 11.5 years. Delayed puberty is defined as absence of testicular enlargement by age 14. The most common cause is self-limited delayed puberty (SLDP), often familial and benign. Functional hypogonadotropic hypogonadism due to chronic illness, and permanent central hypogonadism (congenital or acquired), account for additional cases. Congenital hypogonadotropic hypogonadism (CHH), including Kallmann syndrome, is frequently genetic, with variants in genes such as FGFR1, ANOS1 and GNRHR. Clinical assessment includes family history, growth patterns, and red flags such as micropenis, cryptorchidism or anosmia.
Keywords: AMH; delayed puberty; FSH; inhibin B; testis